: diagnóstico, tratamento e mais!")
A esclerose lateral amiotrófica (ELA) é uma doença neurodegenerativa que afeta o neurônio motor, causando principalmente fraqueza muscular progressiva e que possui péssimo prognóstico. Confira agora os principais aspectos referentes a esta condição neurológica, que aparecem nos atendimentos e como são cobrados nas provas de residência médica!
Navegue pelo conteúdo
Dicas do Estratégia para provas
Seu tempo é precioso e sabemos disso. Se for muito escasso neste momento, veja abaixo os principais tópicos referentes à ELA.
- É uma doença neurodegenerativa que resulta na degeneração seletiva dos neurônios motores superior e inferior.
- A síndrome do neurônio motor superior inclui hipertonia elástica, hiperreflexia e sinal de Babinski.
- A síndrome do neurônio motor inferior os reflexos estão hipoativos, ocorre hipotonia ou atrofia dos músculos e fasciculações
- A ELA é um diagnóstico de exclusão dentro da fraqueza muscular progressiva.
- O único tratamento disponível para ELA esporádica inclui a o Riluzol, onde ensaios clínicos demonstraram retardar discretamente a progressão da doença.
Definição da doença
A esclerose lateral amiotrófica (ELA) foi descrita pela primeira vez por Charcot no século XIX, é uma doença neurodegenerativa que resulta na degeneração seletiva dos neurônios motores superior e inferior. É um dos diagnósticos diferenciais de fraqueza muscular progressiva.
Há um subtipo hereditário, com início mais precoce, chamado de Atrofia Muscular Espinhal (AME), tem herança recessiva bem conhecida, relacionada ao gene SMN.
Epidemiologia e fisiopatologia da ELA
A ELA pode ser classificada em esporádica (90-95% dos casos) ou familiar (5 a 10%). A prevalência estimada é de 5 casos a cada 100 mil habitantes, mas diagnosticadas em apenas 1 a 2 casos por 100 mil habitantes, o que reflete a rápida letalidade da doença.
Os únicos fatores de risco estabelecidos para ELA são a idade e o histórico familiar. A incidência aumenta a cada década, especialmente após os 40 anos de idade, e atinge o pico na sétima década de vida. Por este motivo, há uma tendência no aumento da sua prevalência devido ao envelhecimento da população em países em desenvolvimento.
Alguns fatores adicionais, mas com evidência mais fraca ou conflitantes incluem tabagismo, exposição a metais pesados, trabalho manual pesado ou exercício físico intenso durante a vida e ocupações socioeconômicas mais altas. Alguns genes que atuam como genes mendelianos na ELA familiar, como SOD1 , C9ORF72 e TARDBP, também podem ser fatores de suscetibilidade de baixa penetrância na ELA esporádica.
A fisiopatologia da ELA é caracterizada por degeneração e morte do neurônio motor com gliose substituindo os neurônios perdidos. Neurônios e células gliais em degeneração mostram inclusões intracelulares. Os achados patológicos podem demonstrar células motoras corticais em degeneração, medula espinhal atrófica e os músculos afetados com atrofia de desnervação.
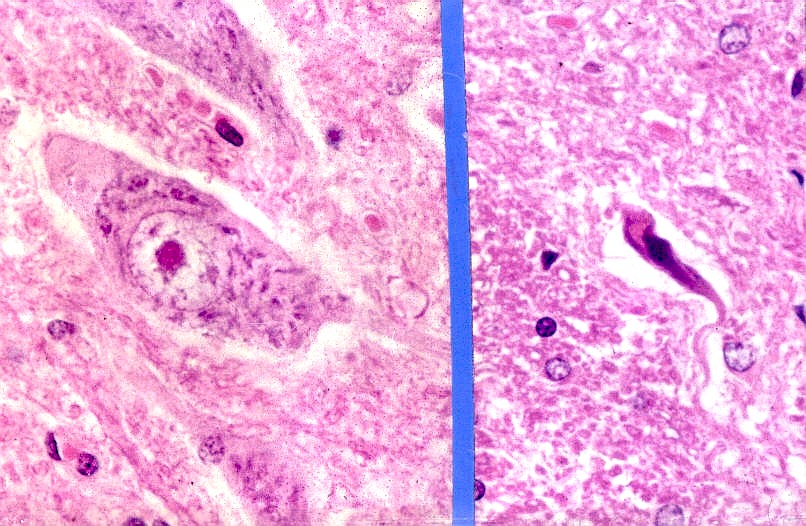

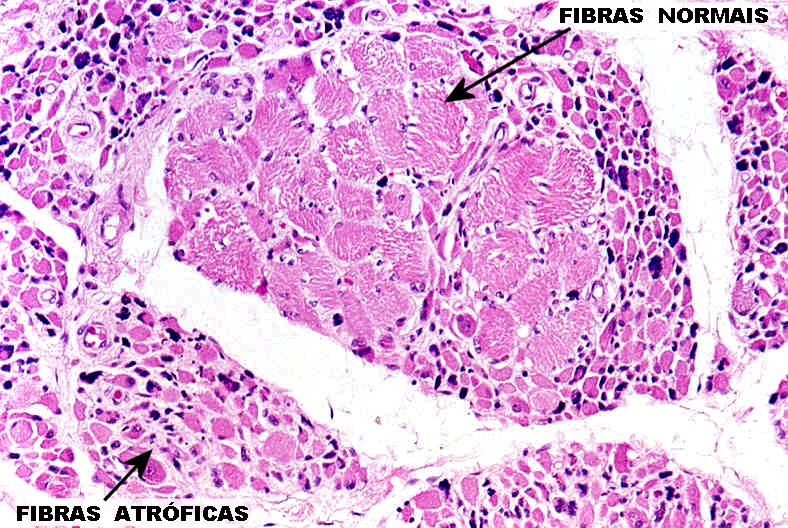
Manifestações clínicas da ELA
A doença se manifesta como fraqueza muscular crônica e assimétrica, com atrofia muscular progressiva, sem alterações sensitivas. De início mais comum acima da quinta década de idade, mas pode ser mais precoce ou tardia. Semelhante à maioria das doenças neurodegenerativas, os sintomas começam de forma focal, com posterior disseminação para outras regiões do corpo.
É importante entender se os sintomas se apresentam como uma síndrome do neurônio motor superior NMS) ou inferior (NMI). No primeiro, ocorre lesão no NMS e liberação do tônus do NMI. Os sinais incluem hipertonia elástica, hiperreflexia e sinal de Babinski.
Disartria e disfagia são os sintomas mais comuns do neurônio motor superior bulbar. Outro sintoma frequente do neurônio motor superior bulbar é a síndrome do afeto pseudobulbar. Isso se manifesta como risadas, choros ou bocejos inapropriados.
Na síndrome do NMI, a lesão periférica os reflexos estão hipoativos, ocorre hipotonia ou atrofia dos músculos e fasciculações.
O curso progressivo da ELA pode eventualmente produzir aspectos da doença que ameaçam a vida, como insuficiência respiratória neuromuscular e disfagia, geralmente como resultado da fraqueza progressiva dos membros e ou dos músculos bulbares.
Diagnóstico de ELA
A ELA é um diagnóstico de exclusão dentro da fraqueza muscular progressiva. Os exames de imagem, como a ressonância magnética, são utilizados para descartar outras causas, principalmente estenose de canal cervical.
A eletroneuromiografia está indicada e faz parte dos critérios diagnósticos. Ela pode revelar características combinadas de desnervação aguda e crônica, mas estes estudos de condução nervosa sensorial e motora são mais frequentemente normais na ELA. Sua importância maior está em comprovar a síndrome do 2° neurônio motor, além de excluir outras neuropatias motoras.
Os critérios revisados da Federação Mundial de Neurologia de El Escorial, também conhecidos como critérios de Airlie House, incluem sinais do neurônio motor superior e inferior e progressão dos sintomas ao longo do tempo.
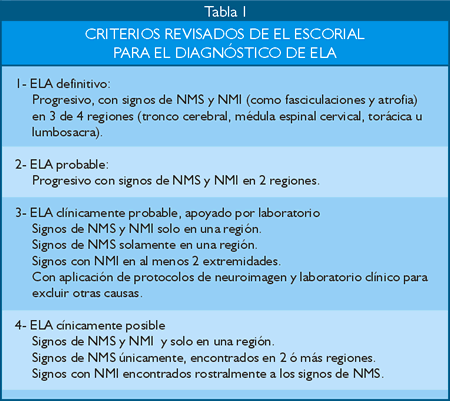
Tratamento da ELA
O prognóstico é ruim na maioria dos casos. O único tratamento disponível para ELA esporádica inclui a o Riluzol, onde ensaios clínicos demonstraram retardar a progressão da doença, aumentam o tempo até a necessidade de traqueostomia ou ventilação assistida permanente ou prolongam a sobrevida. A administração do Riluzol é feita por comprimido, suspensão ou filme de desintegração oral, cada um na dose de 50 mg duas vezes ao dia.
No caso da Atrofia Muscular Espinhal (AME), o tratamento foi revolucionado recentemente com o uso de Nusinersen, agindo diretamente na alteração genética do gene SMN.
É importante uma abordagem multiprofissional com a participação de fonoaudiologia e fisioterapia para reabilitação motora. Os pacientes que evoluem com insuficiência respiratória podem precisar de suporte ventilatório, inclusive traqueostomia. Cuidados paliativos são indicados para pacientes com doença avançada.

Veja também:
- Cérebro: anatomia, funções e muito mais
- Resumo de coma e alteração da consciência
- Resumo de doenças da coluna vertebral
- Resumo de cefaleias: primária, secundária, abordagens e muito ma
- Resumo de AVCi
- Neurocirurgia: sistema nervoso, doenças e mais
- Neurologia: o que é, doenças, residência médica, salários e mais
- Demência: o que é, sintomas e muito mais
- Alzheimer: o que é, sintomas, diagnóstico e tratamento
Referências bibliográficas:
- UNICAMP, Universidade Estadual de Campinas . Anatpat: Anatomia Patológica para Graduação – Peças e Lâminas.Disponível em: Anatpat, Unicamp.
- Chieia, Marco A. et al. Esclerose lateral amiotrófica: considerações sobre critérios diagnósticos. Arquivos de Neuro-Psiquiatria [online]. 2010, v. 68, n. 6, pp. 837-842. Disponível em: https://doi.org/10.1590/S0004-282X2010000600002
- Júnior, Mário Emílio Teixeira Dourado. Estudo epidemiológico da Esclerose Lateral Amiotrófica no Brasil. UFRN. Natal-RN, 2020.
- Oliveira and Pereira. Amyotrophic lateral sclerosis. Arq Neuropsiquiatr 2009
- Clinical features of amyotrophic lateral sclerosis and other forms of motor neuron disease. Uptodate.
- Crédito da imagem em destaque: Pixabay


