
Olá, meu Doutor e minha Doutora! O termo “amiloide” foi introduzido pela primeira vez por Schleiden em 1838 para descrever o amido vegetal, adotado por Rudolf Virchow em 1854 para descrever depósitos teciduais que coravam de maneira semelhante à celulose quando expostos ao iodo.
Vem com o Estratégia MED entender o que são os depósitos amiloides e como se manifesta no paciente com amiloidose. Ao final, separei uma questão de prova sobre o tema para respondermos juntos!
Navegue pelo conteúdo
Conceito de Amiloidose
A amiloidose é caracterizada pela deposição de fibrilas altamente organizadas de proteínas em tecidos extracelulares, as quais podem levar a diversas manifestações clínicas. Essas fibrilas são compostas por subunidades de baixo peso molecular de várias proteínas que, em sua forma normal, circulam no plasma. A formação desses depósitos amiloides resulta da alteração conformacional de peptídeos precursores solúveis, que adotam uma configuração de folha beta-plissada, permitindo que se agrupem em protofilamentos fibrilares torcidos.
Conheça o curso mais completo para conquistar sua vaga na Residência Médica!
Prepare-se com o Estratégia MED!
Patogênese da Amiloidose
Os depósitos amiloides consistem em fibrilas insolúveis compostas por subunidades proteicas de baixo peso molecular. Essas subunidades, derivadas de precursores solúveis, sofrem mudanças conformacionais resultantes na adoção de uma configuração predominantemente antiparalela de folhas beta-plissadas, levando à formação de fibrilas altamente organizadas.
Diversos fatores influenciam a fibrilogênese, incluindo alterações genéticas, enovelamento e estabilidade de proteínas, além da presença de componentes não-fibrilares, como glicosaminoglicanos e apolipoproteínas.
A amiloidose pode ser de origem genética, com mutações em várias proteínas precursoras associadas a diferentes tipos de amiloidose. As doenças amiloides hereditárias podem resultar de mutações que tornam as proteínas mais propensas à agregação e fibrilogênese, polimorfismos em cofatores ou proteínas de subunidades, e doenças que afetam o nível ou acumulação de proteínas precursoras. A maioria das formas hereditárias de amiloidose é dominante e heterozigota, com ambas as moléculas selvagens e mutantes presentes nos depósitos amiloides.
Tipos de Amiloidose
Existem várias formas de amiloidose sistêmica, com 18 tipos diferentes e 28 formas localizadas. Os tipos sistêmicos mais comuns são os primários (AL) e a amiloidose associada à transtirretina (ATTR), mas também há outros tipos clinicamente relevantes, como a amiloidose secundária (AA). A nomenclatura das proteínas precursoras inclui a letra “A” seguida da abreviação do nome da proteína precursora. Os principais tipos e suas características são:
- Amiloidose AL: resulta da deposição de proteínas derivadas de fragmentos de cadeias leves de imunoglobulinas, geralmente associada a discrasias de células plasmáticas.
- Amiloidose ATTR: pode ser classificada como selvagem (ATTRwt), associada ao envelhecimento, ou mutante (ATTRv ou hATTR), associada à neuropatia familiar e/ou cardiomiopatia.
- Amiloidose AA: complicação de doenças crônicas com inflamação contínua, como artrite reumatoide, resultando na produção sustentada de proteína amiloide A sérica.
- Amiloidose relacionada à diálise: ocorre em pacientes com doença renal terminal mantidos por longos períodos na diálise devido à deposição de fibrilas de beta2-microglobulina.
- Amiloidoses hereditárias: causadas por mutações genéticas que levam à produção de proteínas precursoras que podem formar fibrilas amiloides.
- Amiloidose órgão-específica: deposição de amiloide em um único órgão, como pele, olho, coração, pâncreas, trato gastrointestinal ou geniturinário, resultando em síndromes específicas.
- Amiloidose iatrogênica: associada ao uso de certos medicamentos terapêuticos que podem levar à formação de depósitos amiloides.
Manifestações Clínicas da Amiloidose
A amiloidose é uma condição complexa com uma ampla gama de manifestações clínicas que variam dependendo do tipo de proteína precursora envolvida, da distribuição do amiloide nos tecidos e da quantidade depositada:
- Envolvimento renal: manifesta-se geralmente como proteinúria ou síndrome nefrótica, mas também pode levar à insuficiência renal avançada em alguns casos. Os tipos AA e AL são comuns, enquanto o tipo ATTR afeta raramente os rins.
- Cardiomiopatia: importante manifestação que pode causar disfunção cardíaca, insuficiência cardíaca, arritmias e angina. A amiloidose cardíaca é comum nos tipos ATTR e AL.
- Trato gastrointestinal: hepatomegalia é comum em alguns tipos de amiloidose. Além disso, sintomas como sangramento, gastroparesia e constipação podem ocorrer devido à deposição amiloide.
- Anormalidades neurológicas: podem incluir neuropatia periférica e autonômica, envolvimento do sistema nervoso central e acidente vascular cerebral isquêmico. A neuropatia periférica é comum em amiloidoses hereditárias e na AL.
- Manifestações musculoesqueléticas: pseudo-hipertrofia muscular, macroglossia, artropatia e outros sintomas podem ocorrer devido à deposição amiloide nos músculos, articulações e tecidos moles periarticulares.
- Manifestações hematológicas: podem incluir sangramento aumentado devido à redução da atividade do fator X, anemia, trombocitopenia e outras manifestações relacionadas ao grau de acometimento do órgão.
- Manifestações pulmonares: incluem infiltração traqueobrônquica, derrames pleurais persistentes e nódulos parenquimatosos.
- Manifestações cutâneas: sinais como pele cerosa e hematomas fáceis são comuns em alguns tipos de amiloidose e podem ser altamente característicos.
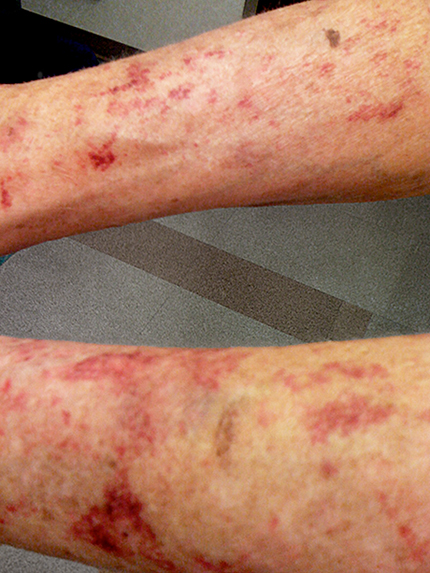
Diagnóstico da Amiloidose
A confirmação do diagnóstico geralmente requer uma biópsia tecidual, embora exames de imagem também possam ser úteis. A escolha do local da biópsia depende dos órgãos afetados e da apresentação clínica. A análise histopatológica e proteica dos depósitos amiloides é crucial para determinar o tipo de amiloide presente.
Para suspeita de amiloidose de cadeia leve (AL), é fundamental investigar uma possível discrasia de células plasmáticas. No entanto, a presença de uma proteína monoclonal isolada não é suficiente para o diagnóstico de amiloidose AL, a menos que as cadeias leves nos depósitos amiloides sejam confirmadas por técnicas específicas.
Exames de imagem, como cintilografia e PET-CT, podem ser valiosos para avaliar a distribuição e a carga de depósitos amiloides, especialmente em casos de suspeita de amiloidose cardíaca.
Tratamento da Amiloidose
O tratamento da amiloidose é altamente variável, dependendo do tipo específico da doença e das condições subjacentes do paciente:
- Amiloidose AL: o tratamento visa suprimir a discrasia de células plasmáticas subjacente.
- Amiloidose AA: a terapia visa suprimir o distúrbio infeccioso ou inflamatório subjacente.
- Amiloidose por Transtirretina (ATTR): várias abordagens estão disponíveis, incluindo terapias-alvo de RNA e estabilização de tetrâmeros transtirretina. O transplante hepático também é uma opção.
- Amiloidose relacionada à diálise: o tratamento envolve mudar o modo de diálise ou considerar o transplante renal.
Existem várias abordagens em investigação, incluindo silenciamento de RNA, edição genética, imunoterapia, direcionamento do componente P amiloide sérico e agentes que interrompem fibrilas. O manejo de pacientes com envolvimento de órgãos isolados depende da região afetada e do tipo de amiloide presente.
Cai na Prova
Acompanhe comigo uma questão sobre o tema (disponível no banco de questões do Estratégia MED):
RJ – SECRETARIA MUNICIPAL DE ADMINISTRAÇÃO DE MACAÉ – SEMAD 2023 Macroglossia, equimoses periorbitárias e distrofia das unhas são sinais clínicos observados em quais das patologias abaixo:
A. Sarcoidose
B. Amiloidose AL
C. Doença do enxerto versus hospedeiro
D. Anemia falciforme.
E. Artrite psoriásica.
Comentário da Equipe EMED: Correta a alternativa B. Nessa forma, alguns achados são muito característicos e, quando descritos em um enunciado, devem chamar sua atenção. São eles: Macroglossia: quando presente, é altamente sugestiva e está associada a lesões na região lateral da língua, pelo contato direto com os dentes. Lesões purpúricas ao redor dos olhos ou “olhos de guaxinim”. Proteinúria e síndrome nefrótica. Cardiomiopatia restritiva: insuficiência cardíaca com seus sinais e sintomas clássicos (p. ex., dispneia, edema) e dissociação massa-voltagem (aumento da massa cardíaca com redução da voltagem no eletrocardiograma); e Neuropatia periférica (p.ex., síndrome do túnel do carpo, polineuropatia sensitiva ou sensitivomotora) e neuropatia autonômica (p.ex., hipotensão ortostática). A distrofia ungueal é descrita, mas é um achado inespecífico.
Venha fazer parte da maior plataforma de Medicina do Brasil! O Estrategia MED possui os materiais mais atualizados e cursos ministrados por especialistas na área. Não perca a oportunidade de elevar seus estudos, inscreva-se agora e comece a construir um caminho de excelência na medicina!
Veja Também
- ResuMED de sarcoidose: amiloidose, doença relacionada à IgG4 e Síndrome de Sapho
- ResuMED de artrite reumatoide: epidemiologia, manifestações, diagnóstico, tratamento e mais!;
- Resumo de artrite reumatoide: manifestações clínicas, diagnóstico, tratamento e mais!;
- Residência Médica em Reumatologia: rotina, remuneração, estudos e mais!;
- Sociedade Brasileira de Reumatologia.


