
Olá, meu Doutor e minha Doutora! A fenilcetonúria pode ser entendida como a presença de fenilalanina e compostos cetônicos nos líquidos corporais, indicando a incapacidade das pessoas com esse distúrbio de metabolizar adequadamente a fenilalanina, levando à sua acumulação e à presença de certos compostos no corpo.
Vem com o Estratégia MED entender como ocorre o processo da fenilcetonúria e como identificamos nos pacientes, principalmente através da triagem neonatal!
Navegue pelo conteúdo
O que é a Fenilcetonúria
A fenilcetonúria (PKU), menos comumente conhecida como deficiência de fenilalanina hidroxilase, é o erro inato mais comum do metabolismo de aminoácidos. A deficiência da enzima fenilalanina hidroxilase (PAH) prejudica a capacidade do corpo de metabolizar o aminoácido essencial fenilalanina, levando ao acúmulo de fenilalanina em fluidos corporais.
Fisiopatologia da Fenilcetonúria
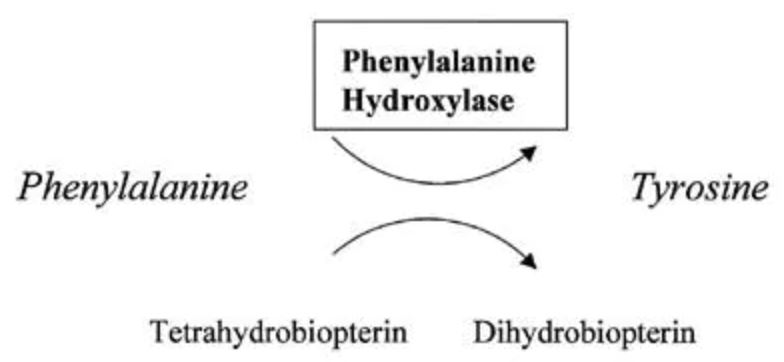
A enzima hepática fenilalanina hidroxilase (PAH) catalisa a conversão do aminoácido essencial fenilalanina em tirosina. A PKU é causada pela deficiência de PAH. Isso resulta em concentrações elevadas de fenilalanina no sangue (>1200 μmol/L) e na urina e seus metabólitos, fenilacetato e fenillactato. A tetraidrobiopterina (BH4) é um cofator também necessário que mantêm a PAH em sua forma ativa. A concentração elevada de fenilalanina causa deficiência neurológica, mas o mecanismo é desconhecido.
Epidemiologia da Fenilcetonúria
A fenilcetonúria é mais comumente diagnosticada em neonatos devido a programas de triagem neonatal. A prevalência de PKU é mais alta em populações europeias e do leste asiático, ocorrendo em aproximadamente 1:10.000 a 1:15.000 nascidos vivos. Alguns países europeus, como a Itália, Finlândia e Irlanda, podem apresentar maior prevalência em comparação com outros.
A maioria dos casos de PKU no mundo é causada pela deficiência de fenilalanina hidroxilase ou, menos comumente, pela deficiência de tetraidrobiopterina (BH4). Em todo o mundo, a PKU é mais comum em brancos e asiáticos.
Deficiência da Enzima fenilalanina Hidroxilase
Etiologia
Quase todos os casos são causados por mutações no gene que codifica a fenilalanina hidroxilase (HAP) sendo autossômico recessivo. Mais de 1000 mutações estão associadas à deficiência de HAP.
Quadro clínico
Devido à triagem neonatal generalizada, manifestações clínicas evidentes de PKU são raras. Os recém-nascidos são assintomáticos antes do início da amamentação contendo fenilalanina (por exemplo, leite materno ou fórmula infantil padrão). Se não for detectada durante o período neonatal, o início da PKU é insidioso e pode não causar sintomas até a primeira infância.
Em pacientes não tratados, a característica da doença é deficiência intelectual irreversível, convulsões, anormalidades comportamentais, microcefalia e doença de pele, como erupção eczematosa e pigmentação leve, devido à hiperfenilalaninemia. O corpo e a urina podem ter um odor devido ao aumento da concentração de ácido fenilacético. Sem restrição alimentar, o comprometimento cognitivo piora durante a mielinização na primeira infância com o aumento da exposição dietética à fenilalanina, mas se estabiliza quando a maturação cerebral está completa.
Diagnóstico
O diagnóstico de PKU é baseado na descoberta de uma concentração sérica elevada de fenilalanina, seguida de teste molecular. O método laboratorial mais útil para a triagem neonatal é a espectrometria de massas em tandem. Este método também pode medir aminoácidos adicionais, incluindo tirosina. Uma alta concentração de fenilalanina juntamente com baixa a baixa concentração normal de tirosina sugere o diagnóstico de PKU.
Tratamento
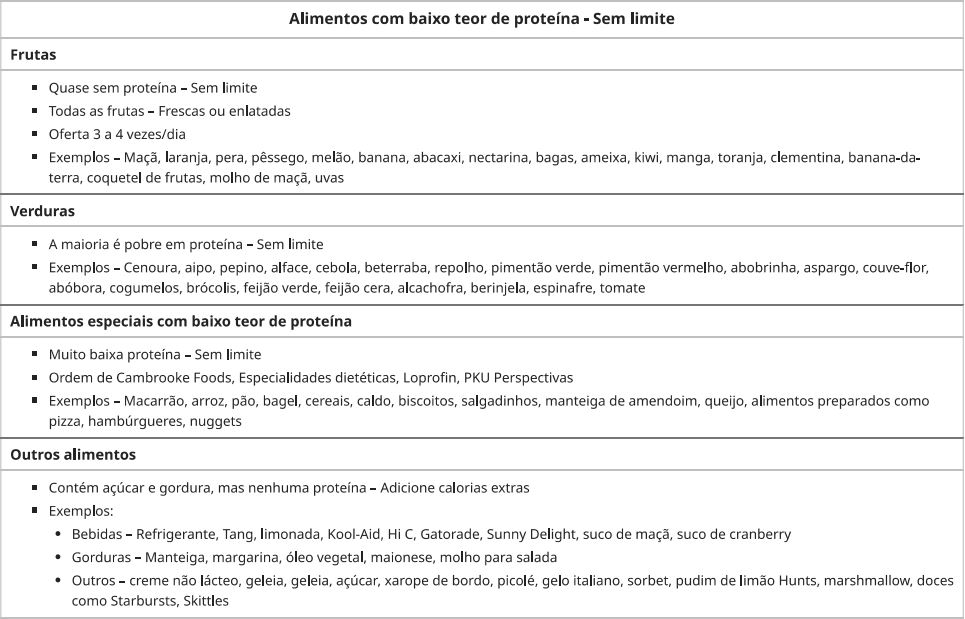
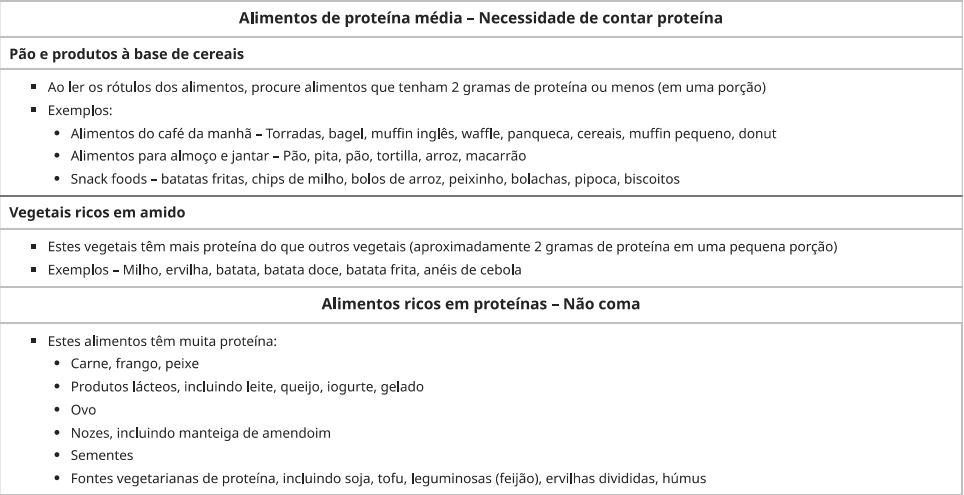
O manejo de pacientes tem como base a restrição dietética de fenilalanina, amamentação alternada com fórmula livre de fenilalanina e farmacoterapia com formulação sintética de BH4 chamada sapropterina.
Deficiência de tetraidrobiopterina (BH4)
Etiologia
A maioria dos distúrbios do metabolismo do BH4 é autossômica recessiva, sendo responsáveis por aproximadamente 2% dos pacientes com níveis elevados de fenilalanina.
Quadro Clínico
Os pacientes afetados geralmente apresentam HAP e/ou deterioração neurológica progressiva durante a infância devido à diminuição da produção dos neurotransmissores dopamina, epinefrina, noradrenalina e serotonina. Os pacientes não tratados geralmente falecem antes de completar um ano.
Diagnóstico
Qualquer recém-nascido com concentrações elevadas de fenilalanina deve ser testado para deficiência de BH4. O diagnóstico é feito pela medição de concentrações elevadas de biopterina ou neopterina no sangue, urina ou líquido cefalorraquidiano.
Tratamento
A deficiência de BH4 é tratada com uma dieta pobre em fenilalanina e suplementação com BH4. Isso diminuirá as concentrações de fenilalanina, mas não melhorará o estado neurológico. Os precursores do neurotransmissor, L-dopa, carbidopa, e serotonina, também são fornecidos.
Venha fazer parte da maior plataforma de Medicina do Brasil! O Estrategia MED possui os materiais mais atualizados e cursos ministrados por especialistas na área. Não perca a oportunidade de elevar seus estudos, inscreva-se agora e comece a construir um caminho de excelência na medicina!

Veja também:
- Teste do pezinho: tudo sobre!
- Resumo de Espinha Bífida: fatores de risco, diagnóstico e mais!
- Resumo de hidrocefalia em crianças: diagnóstico, tratamento e mais!
- Resumo de convulsão febril: fisiopatologia, classificação, profilaxia
- Resumo de choque em pediatria: conceitos, fisiopatologia e classificação
- Resumo sobre laringite aguda: diagnóstico, tratamento e mais!
- Resumo de infecção urinária em pediatria


