
Olá, querido doutor e doutora! A Síndrome de Morris, também conhecida como Síndrome de Insensibilidade Androgênica Completa (CAIS), é uma condição genética rara que afeta o desenvolvimento sexual em indivíduos com cariótipo XY. Devido a mutações no gene que codifica o receptor de andrógeno, o corpo é incapaz de responder aos hormônios masculinos, resultando em características físicas femininas, apesar da presença de cromossomos típicos do sexo masculino. Ao longo deste texto, serão explorados os aspectos fisiopatológicos, o quadro clínico, o diagnóstico e as opções de tratamento para essa condição.
Navegue pelo conteúdo
Conceito de Síndrome de Morris
A Síndrome de Morris, também conhecida como síndrome de insensibilidade androgênica completa (CAIS), é uma condição genética rara caracterizada pela incapacidade das células em responder adequadamente aos andrógenos, os hormônios sexuais masculinos. Apesar da presença de cromossomos XY, típicos do sexo masculino, os indivíduos com esta síndrome desenvolvem características físicas femininas, como ausência de pelos corporais, desenvolvimento mamário e genitália externa feminina, devido à insensibilidade aos efeitos dos andrógenos. Essa condição resulta em um fenótipo feminino em indivíduos geneticamente masculinos, com testículos geralmente não descendidos e ausência de útero e trompas de Falópio.
Fisiopatologia da Síndrome de Morris
A fisiopatologia da Síndrome de Morris envolve uma mutação no gene AR (receptor de andrógeno) localizado no cromossomo X, que codifica o receptor de andrógeno, uma proteína importante para a ação dos hormônios androgênicos. Em condições normais, os andrógenos, como a testosterona, se ligam a esses receptores, que então ativam a transcrição de genes responsáveis pelo desenvolvimento das características sexuais masculinas. Na Síndrome de Morris, as mutações no gene AR resultam em um receptor de andrógeno não funcional ou ausente, impedindo a ligação adequada dos andrógenos.
Devido à falta de resposta das células aos andrógenos, o desenvolvimento das características sexuais masculinas é interrompido, apesar da presença de níveis normais ou elevados de testosterona e outros andrógenos. O corpo, então, segue o caminho de desenvolvimento padrão feminino, explicando a presença de características femininas, como genitália externa feminina, e ausência de estruturas internas, como útero e ovários. Os testículos, apesar de presentes, geralmente permanecem intra-abdominais ou inguinais e não descem para o escroto, levando ao risco aumentado de neoplasias germinativas testiculares.
Epidemiologia e Fatores de Risco da Síndrome de Morris
A Síndrome de Morris é uma condição genética rara, com uma incidência estimada entre 1 em 20.000 a 1 em 64.000 nascidos do sexo masculino. A síndrome afeta indivíduos com cariótipo XY, que, devido à mutação no gene AR no cromossomo X, apresentam características fenotípicas femininas. Como a herança é ligada ao cromossomo X, a condição é geralmente transmitida de mãe para filho, sendo que a mãe é uma portadora assintomática.
Os fatores de risco para a Síndrome de Morris estão diretamente relacionados à história familiar de insensibilidade androgênica. Mulheres portadoras da mutação AR têm 50% de chance de transmitir a mutação para seus filhos, que desenvolverão a síndrome se forem geneticamente masculinos (XY). Não há fatores de risco ambientais ou comportamentais conhecidos que contribuam para o desenvolvimento dessa condição, dado seu caráter exclusivamente genético. A condição pode se manifestar de maneira esporádica em casos de mutações de novo, sem histórico familiar prévio.
Quadro Clínico da Síndrome de Morris
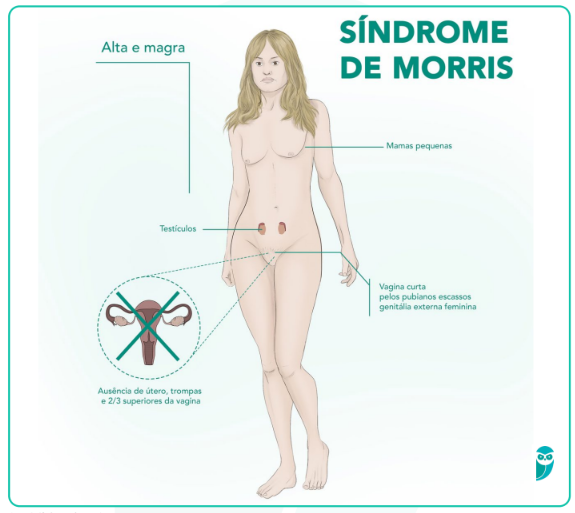
A Síndrome de Morris apresenta um quadro clínico característico que reflete a incapacidade do organismo de responder aos hormônios androgênicos, apesar da presença de cromossomos XY. Embora os indivíduos afetados tenham um cariótipo masculino, o desenvolvimento físico segue um padrão feminino, o que frequentemente leva à descoberta da condição durante a adolescência, quando ocorrem discrepâncias no desenvolvimento sexual secundário. A ausência de menstruação é o sinal mais comum que leva à investigação e diagnóstico da síndrome, revelando uma série de características clínicas específicas:
- Genitália externa feminina normal;
- Desenvolvimento mamário completo durante a puberdade;
- Amenorreia primária;
- Ausência de pêlos pubianos e axilares;
- Ausência de útero, trompas de Falópio e ovários;
- Presença de testículos intra-abdominais ou inguinais;
- Vagina em fundo cego;
- Estatura superior à média; e
- Aumento do risco de malignidade testicular.
Diagnóstico da Síndrome de Morris
Para confirmar o diagnóstico, são realizados exames laboratoriais que revelam níveis elevados de testosterona, hormônio luteinizante (LH) e hormônio folículo-estimulante (FSH), indicando uma insensibilidade aos andrógenos. O cariótipo é fundamental para identificar a presença de cromossomos XY em indivíduos com fenótipo feminino.
A confirmação definitiva é obtida por meio de testes genéticos que detectam mutações no gene AR, responsável pela codificação do receptor de andrógeno. Em alguns casos, pode ser necessária uma biópsia testicular para avaliar o risco de malignidade.
Tratamento da Síndrome de Morris
O tratamento da Síndrome de Morris é multidisciplinar e visa principalmente o manejo das complicações associadas e a orientação sobre o desenvolvimento sexual e reprodutivo. Uma das principais considerações no tratamento é a remoção dos testículos ectópicos, que têm um risco aumentado de malignidade ao longo da vida. A orquiectomia, geralmente realizada após a puberdade, é recomendada para minimizar esse risco. Após a remoção dos testículos, a terapia de reposição hormonal com estrogênio é indicada para manter as características femininas secundárias, como o desenvolvimento mamário e a saúde óssea.
Além das intervenções cirúrgicas e hormonais, o suporte psicológico é fundamental, pois o diagnóstico pode ter um impacto significativo na identidade de gênero e na saúde mental das pacientes. Aconselhamento genético também é recomendado para informar a paciente e sua família sobre a natureza hereditária da condição e as implicações para futuras gerações. Em casos onde a paciente deseja ter uma vida sexual ativa, pode ser necessário realizar procedimentos cirúrgicos para alongar a vagina (vaginoplastia), dependendo da anatomia individual.
Cai na Prova
Acompanhe comigo uma questão sobre o tema (disponível no banco de questões do Estratégia MED):
SP – Universidade de Taubaté – Unitau (Hospital Universitário de Taubaté) 2024, 16 anos, procura o serviço de ginecologia devido quadro de amenorréia primária. Nega vida sexual. Nega dor. Ao exame físico, caracteres sexuais e somáticos aparentemente normais e compatíveis, genitália externa de aspecto normal. Assinale a alternativa mais adequada ao caso:
A. Hipótese de hímen imperfurado, solicitar exame de imagem da pelve.
B. Hipótese de síndrome de Turner, indicado cariótipo, imagem da pelve e perfil hormonal.
C. Hipótese de síndrome de Morris, solicitar cariótipo, imagens da pelve e perfil hormonal.
D. Hipótese de má-formação dos dutos mesonéfricos, solicitar exame de imagem da pelve.
Comentário da Equipe EMED: Alternativa “c” correta, pois na síndrome de Morris há uma amenorreia com caracteres sexuais secundários desenvolvidos, porém sem desenvolvimento de útero, terço superior de vagina e tubas.
Venha fazer parte da maior plataforma de Medicina do Brasil! O Estrategia MED possui os materiais mais atualizados e cursos ministrados por especialistas na área. Não perca a oportunidade de elevar seus estudos, inscreva-se agora e comece a construir um caminho de excelência na medicina!
Veja Também
- Resumo de amenorreia: manifestações clínicas, diagnóstico, tratamento e mais!
- Resumo de Anatomia e embriologia do sistema reprodutor feminino
- Resumo de dor pélvica crônica e Dismenorreia: fisiopatologia, diagnóstico, tratamento e mais
- Puerpério: o que é e muito mais
- Candidíase: o que é, tipos e muito mais
- Resumo sobre Clamídia: manifestações clínicas, diagnóstico, tratamento e mais
- Gonorreia: sintomas, tratamentos e muito mais
- Saúde da Mulher: importância, doenças e mais
- Mastologia: rotina, salário residência médica e mais
Referências Bibliográficas
- UHLIG, Rebecca. Androgen Insensitivity Syndrome. In: STATPEARLS. Treasure Island (FL): StatPearls Publishing, 2023. Disponível em: https://www.ncbi.nlm.nih.gov/books/NBK542206/. Acesso em: 2 set. 2024.


