E aí, doc! Vamos explorar mais um tema essencial? Hoje o foco é a Síndrome Hemofagocítica, uma condição inflamatória grave causada por uma ativação descontrolada do sistema imune, que pode evoluir rapidamente e exige diagnóstico e manejo ágeis.
O Estratégia MED está aqui para descomplicar esse conceito complexo e ajudar você a aprofundar seus conhecimentos, promovendo uma prática clínica cada vez mais precisa e segura.
Vamos nessa!
Navegue pelo conteúdo
Definição de Síndrome Hemofagocítica
A síndrome hemofagocítica é uma condição grave, rara e frequentemente fatal, que resulta de uma ativação desregulada e exacerbada do sistema imune. Essa ativação excessiva leva a uma produção descontrolada de citocinas inflamatórias, um fenômeno conhecido como tempestade de citocinas, que culmina em um estado hiperinflamatório sistêmico, podendo causar disfunção e falência de múltiplos órgãos.
A síndrome pode ter origem hereditária, quando há mutações genéticas que comprometem a função de células imunológicas, ou pode ser adquirida, associada a infecções graves, doenças autoimunes ou neoplasias.
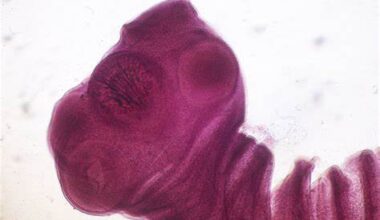
Clinicamente, os pacientes geralmente apresentam febre persistente, citopenias (redução das células do sangue), hepatoesplenomegalia (aumento do fígado e do baço) e sinais laboratoriais de inflamação intensa. A hemofagocitose, fagocitose de células sanguíneas por macrófagos ativados, pode ser identificada em amostras da medula óssea, baço ou linfonodos.
Historicamente, os primeiros relatos da síndrome datam de 1952, com a descrição de dois irmãos com febre e hepatoesplenomegalia, sugerindo um padrão familiar da doença. Desde então, casos esporádicos e familiares foram documentados, levando a um crescente interesse na sua compreensão.
Etiologia da Síndrome
A etiologia da síndrome hemofagocítica pode ser classificada em duas formas principais: primária (ou familiar) e secundária (ou adquirida), sendo ambas marcadas por uma ativação imunológica descontrolada, mas com causas distintas.
Síndrome hemofagocítica primária
A forma primária é de origem genética e geralmente se manifesta na infância, embora existam relatos em adultos. Está relacionada a mutações em genes que afetam a função citotóxica dos linfócitos T e das células NK, essenciais para o controle de infecções e células anormais.
Entre os genes mais frequentemente implicados está o PRF1, responsável pela produção da perforina, uma proteína crucial na destruição de células-alvo. Outros genes envolvidos participam da formação, transporte e exocitose dos grânulos citotóxicos. A herança segue padrão mendeliano recessivo. Essa disfunção imunológica congênita impede a regulação adequada da resposta inflamatória, resultando em um estado hiperinflamatório persistente.
Síndrome hemofagocítica secundária
A forma secundária pode ocorrer em qualquer idade, com média de diagnóstico por volta dos 50 anos, e está associada a condições adquiridas que atuam como gatilhos do quadro inflamatório. Os principais fatores desencadeantes são:
- Infecções (principal causa): especialmente virais, como vírus Epstein-Barr (EBV) e citomegalovírus (CMV), mas também podem envolver hepatite A, influenza H1N1, HIV (principalmente em fases avançadas ou após início de tratamento antirretroviral), além de bactérias, fungos e parasitas, geralmente em indivíduos imunocomprometidos ou com histórico de viagens para áreas endêmicas.
- Neoplasias: particularmente linfomas de células T (como o linfoma NK/T nasal), mas também em linfomas de células B, mieloma múltiplo, leucemias e linfoma de Hodgkin. Nesses casos, a síndrome pode ser a primeira manifestação da doença oncológica ou indicar progressão ou recaída.
- Doenças autoimunes: também chamada de síndrome de ativação macrofágica nesse contexto. É mais comum na doença de Still do adulto e lúpus eritematoso sistêmico, podendo também ocorrer em artrite reumatoide, vasculites e doença inflamatória intestinal. Em muitos casos, há coexistência de múltiplos gatilhos, como infecção, parto ou uso de imunossupressores.
- Outras causas: incluem transplantes (especialmente de sangue do cordão umbilical ou rim), gravidez e pós-parto (devido a alterações fisiológicas na atividade das células NK), uso de certos medicamentos como a lamotrigina, e em cerca de 10% dos casos não se identifica nenhuma causa clara.
Além disso, estudos recentes sugerem que a distinção entre formas primária e secundária pode ser artificial em alguns casos, já que pacientes com formas ditas adquiridas podem ter alterações genéticas ainda não identificadas, contribuindo para sua suscetibilidade à síndrome.
Fisiopatologia da Síndrome Hemofagocítica
A síndrome hemofagocítica (SHF) é uma condição grave caracterizada por uma ativação descontrolada do sistema imune, principalmente envolvendo linfócitos T citotóxicos e macrófagos, resultando em inflamação sistêmica intensa e falência multiorgânica. A fisiopatologia varia entre suas formas primária (familiar/genética) e secundária (adquirida), mas ambas compartilham mecanismos comuns de hiperativação imune.
Ativação imunológica normal
Em indivíduos saudáveis, células infectadas por vírus ou transformadas (como células tumorais) apresentam antígenos via complexo de histocompatibilidade classe I, sendo reconhecidas e eliminadas por linfócitos T citotóxicos e células NK. Essas células citotóxicas eliminam os alvos por meio da liberação de perforinas (que formam poros na membrana celular) e granzimas (que entram na célula e ativam a cascata de caspases, promovendo apoptose). A ativação é controlada, com liberação de citocinas inflamatórias como IFN-γ e citocinas reguladoras como IL-10, que equilibram a resposta.
Fisiopatologia da SHF primária
Na forma primária, geralmente associada a mutações genéticas (como nos genes PRF1 e UNC13D), há falência na função citolítica de células T e NK. Essas células não conseguem eliminar efetivamente os alvos, resultando em estimulação prolongada do sistema imune. A exposição persistente de antígenos leva à ativação sustentada de linfócitos T, que passam a produzir altos níveis de IFN-γ e outras citocinas inflamatórias. O defeito genético também pode envolver a maquinaria de exocitose dos grânulos citotóxicos, impedindo a apoptose das células-alvo.
Fisiopatologia da SHF secundária
Na forma secundária, desencadeada por infecções, doenças autoimunes, neoplasias ou medicamentos, há uma hiperestimulação imune adquirida, sem mutações genéticas evidentes na maioria dos casos. O estímulo inicial provoca ativação excessiva dos linfócitos T citotóxicos, que produzem IFN-γ em grandes quantidades.
Esse interferon ativa os macrófagos, levando à hemofagocitose (fagocitose de células sanguíneas), resultando em citopenias. O IFN-γ também promove diferenciação anormal de precursores hematopoiéticos, agravando a anemia.
Além do IFN-γ, há liberação de múltiplas citocinas pró-inflamatórias (como TNF-α, IL-1, IL-6, IL-18) que geram a tempestade de citocinas, responsável por febre persistente, hepatomegalia, hiperferritinemia e disfunção de múltiplos órgãos. A via JAK/STAT é ativada em excesso, intensificando ainda mais a resposta inflamatória.
Outros mecanismos fisiopatológicos
- IL-6: contribui para inflamação crônica, inibe função de células NK e reduz níveis de perforinas e granzimas.
- HO-1: enzima envolvida na degradação do heme, está aumentada na SHF devido à hemofagocitose; seus produtos (biliverdina, monóxido de carbono, ferro) têm efeitos anti-inflamatórios e explicam achados laboratoriais como hiperferritinemia e elevação da bilirrubina.
- SHF pós-quimioterapia: decorre da lise tumoral, com liberação de citocinas inflamatórias e mudança do perfil de resposta imune para Th1, potencializando o risco em indivíduos predispostos.
Manifestações clínicas da Síndrome Hemofagocítica
A síndrome hemofagocítica (SHF) apresenta-se, na maioria dos casos, de forma aguda ou subaguda, com sintomas que geralmente se instalam ao longo de 1 a 4 semanas. A apresentação clínica é bastante inespecífica e pode ser confundida com outras condições infecciosas ou inflamatórias, o que dificulta o diagnóstico precoce. Em pacientes com doenças de base, é fundamental distinguir os novos achados clínicos daqueles previamente existentes.
Principais manifestações clínicas
- Febre persistente: está presente em mais de 90% dos casos e é um dos sinais mais característicos.
- Astenia e perda de peso: sintomas constitucionais frequentes, com impacto significativo no estado geral.
- Esplenomegalia e hepatomegalia: ocorrem em até 86% e 67% dos casos, respectivamente, devido à ativação e proliferação de macrófagos e linfócitos T.
- Linfadenopatia periférica: menos comum, presente em aproximadamente um terço dos pacientes.
Achados laboratoriais
- Citopenias: acometem geralmente duas ou mais linhagens hematológicas, sendo trombocitopenia e anemia mais frequentes que leucopenia. Podem resultar da hemofagocitose e da supressão medular.
- Hiperferritinemia: um dos achados mais marcantes, relacionada à ativação de macrófagos e à inflamação sistêmica.
- Hipofibrinogenemia: comum, podendo estar relacionada à fibrinólise aumentada e disfunção hepática.
- Elevação de transaminases, bilirrubina, LDH e tempo de protrombina: indicam disfunção hepática e inflamação hepática, muitas vezes associadas à IL-1β.
- Hipertrigliceridemia: causada pela inibição da lipoproteína lipase pelo TNF-α.
- Elevação do CD25 solúvel (sIL-2R): indicativo da ativação persistente de linfócitos T.
- Função das células NK reduzida ou ausente.
Envolvimento de órgãos e sistemas
- Sistema nervoso central: acometido em até 31% dos adultos, com manifestações como alteração do nível de consciência, cefaleia, convulsões e sintomas psiquiátricos. Alterações em TC/MRI são comuns.
- Manifestações mucocutâneas: ocorrem em cerca de 25% dos casos, com exantema morbiliforme, pioderma gangrenoso, púrpura, entre outros. Nódulos subcutâneos do tipo paniculite podem sugerir associação a linfomas.
- Distúrbios da coagulação: presentes em até 85% dos casos, com coagulação intravascular disseminada (CIVD) em até metade dos pacientes graves, podendo evoluir com hemorragia severa.
- Comprometimento renal: lesão renal aguda ocorre em cerca de 50% dos pacientes, podendo evoluir para doença renal crônica.
- Comprometimento pulmonar: presente em até metade dos casos, com sintomas como tosse e dispneia. Radiografias podem mostrar derrame pleural, infiltrados e opacidades.
- Sintomas gastrointestinais: comuns e inespecíficos, incluem dor abdominal, alterações do trânsito intestinal e hemorragia digestiva.
Diagnóstico da Síndrome Hemofagocítica
O diagnóstico da síndrome hemofagocítica (SHF) é desafiador, pois não há sinais ou exames patognomônicos. A hemofagocitose — fagocitose de células sanguíneas por macrófagos — pode ser observada em outras condições inflamatórias intensas, como sepse, malária e doenças reumatológicas, o que compromete sua especificidade. Além disso, nem todos os pacientes com SHF apresentam hemofagocitose nos tecidos analisados, como medula óssea, baço ou linfonodos. Ainda assim, o aspirado de medula óssea é indicado para avaliar a presença do fenômeno e excluir neoplasias hematológicas.
Critérios HLH-2004
O sistema HLH-2004 é amplamente utilizado e define SHF com base na presença de cinco dos oito critérios abaixo, ou por confirmação de mutação genética associada à forma familiar:
- Febre persistente
- Esplenomegalia
- Citopenia em pelo menos duas linhagens (anemia, leucopenia, trombocitopenia)
- Hipertrigliceridemia e/ou hipofibrinogenemia
- Presença de hemofagocitose em medula óssea, baço ou linfonodos
- Atividade reduzida ou ausente de células NK
- Hiperferritinemia
- Elevação do receptor solúvel da interleucina-2 (sIL-2R ou CD25)
Embora úteis, esses critérios foram baseados em populações pediátricas, e sua aplicabilidade em adultos ainda levanta dúvidas. Por exemplo, o valor de ferritina >500 µg/L, recomendado pelo HLH-2004, é pouco específico em adultos.
Para essa faixa etária, valores acima de 6000 a 16000 µg/L têm maior relevância diagnóstica. O sIL-2R elevado também é um marcador sensível, e seu rácio com a ferritina pode ajudar a indicar causas malignas da SHF.
HScore
Como alternativa, o HScore, desenvolvido em 2014, foi elaborado especificamente para avaliar SHF secundária em adultos. Ele considera variáveis clínicas, laboratoriais e citológicas:
- Clínicas: febre, organomegalia, imunossupressão
- Laboratoriais: ferritina, triglicerídeos, AST, fibrinogênio, citopenias
- Citológicas: hemofagocitose na medula óssea
O escore total varia entre 0 e 337. Um ponto de corte de 169 oferece sensibilidade e especificidade adequadas, classificando corretamente cerca de 90% dos casos.
Tratamento da Síndrome Hemofagocítica
O tratamento da síndrome hemofagocítica tem como principal objetivo conter a ativação imunológica descontrolada e evitar a falência de órgãos. A abordagem varia conforme a gravidade, a causa subjacente e a resposta à terapia inicial.
Tratamento de primeira linha
Os corticosteroides em alta dose (como metilprednisolona ou dexametasona) e as imunoglobulinas intravenosas (IVIG) são geralmente utilizados de forma imediata, com ação anti-inflamatória e imunomoduladora. Esse esquema busca controlar rapidamente a resposta inflamatória exagerada.
Em situações mais graves, especialmente com envolvimento do sistema nervoso central, podem ser utilizados esquemas baseados nos protocolos HLH-94 ou HLH-2004, incluindo drogas como etoposido e, em alguns casos, metotrexato intratecal. No entanto, a eficácia desses esquemas é maior em crianças do que em adultos, que apresentam menor taxa de resposta e maior toxicidade.
Tratamento específico conforme a causa
- Síndrome de ativação macrofágica (SAM): o anaquinra, um inibidor do receptor de interleucina-1, tem se mostrado eficaz, especialmente em doenças autoimunes. Em casos refratários ou com envolvimento neurológico, pode ser necessário associar etoposido.
- Causas infecciosas: além da imunossupressão, é essencial tratar a infecção subjacente com antimicrobianos adequados. O rituximabe tem papel específico em casos associados ao vírus Epstein-Barr (EBV), por sua capacidade de reduzir a carga viral.
- Causas neoplásicas: a escolha entre tratar primeiro a inflamação ou a neoplasia depende do estado clínico. Quando a síndrome hemofagocítica causa instabilidade, prioriza-se o controle da inflamação, com corticosteroides, IVIG e etoposido, antes de iniciar a quimioterapia.
Casos refratários
Pacientes que não respondem à terapia padrão após 2–3 semanas são considerados refratários. Nesses casos, pode ser adotado o esquema DEP (doxorrubicina lipossomal, etoposido e metilprednisolona), que mostrou boa taxa de resposta em estudos.
Tratamentos emergentes
Novas terapias vêm sendo estudadas para casos refratários ou intolerantes aos tratamentos convencionais:
- Emapalumab: anticorpo monoclonal que bloqueia o IFN-γ, aprovado para síndrome hemofagocítica primária, embora os estudos em adultos ainda estejam em andamento.
- Ruxolitinib: inibidor das JAK1/JAK2, com resultados promissores em modelos experimentais e em ensaios clínicos iniciais.
- Alemtuzumab: anticorpo anti-CD52 utilizado em protocolos pediátricos e em investigação para uso em adultos com leucemia associada à síndrome hemofagocítica.
Sua trajetória na Medicina merece uma base sólida. Com o Estratégia MED, você tem acesso a conteúdos atualizados e ensino de alto nível. Faça parte dessa comunidade e prepare-se para conquistar seus objetivos.

Veja também!
- Resumo de anemia falciforme: diagnóstico, tratamento e mais!
- Resumo de anemia megaloblástica: diagnóstico, tratamento e mais!
- Resumo sobre Leucócitos: o que são, como avaliar e mais!
- Resumo sobre Eritrograma: definição, componentes sanguíneos e mais!
- Resumo sobre Transfusão Sanguínea: tipos, indicações e mais!
Referências
Kenneth L McClain, MD, PhDOlive Eckstein, MDPaul La Rosée, MD. Clinical features and diagnosis of hemophagocytic lymphohistiocytosis. UpToDate, 2025. Disponível em: UpToDate
AFONSO, Guilherme Augusto Sereno Roxo. Síndrome Hemofagocítica no Adulto. 2020. Dissertação (Mestrado Integrado em Medicina) – Instituto de Ciências Biomédicas Abel Salazar, Universidade do Porto, Porto, 2020.