A síndrome de Edwards é caracterizada por anomalias multissistêmicas, representando a segunda síndrome genética após a síndrome de Down. Confira os principais aspectos referentes a esta síndrome que podem ser cobrados nas provas de residência médica!
Dicas do Estratégia para provas
Seu tempo é precioso e sabemos disso. Se for muito escasso neste momento, veja abaixo os principais tópicos referentes à síndrome de Edwards.
- Esta síndrome genética resulta de uma cópia extra do cromossomo 18, que pode ser completa, parcial e em mosaico;
- As manifestações clínicas incluem alterações neurológicas, anormalidades de crescimento, crânio e face, tórax e abdome, extremidades, órgãos genitais, pele e fâneros, além de malformações de órgãos internos;
- A triagem de cromossomopatias começa na USG morfológica do 1° trimestre, em busca do aumento da translucência nucal;
- Após o nascimento, o diagnóstico da SE é usualmente confirmado por meio do estudo cromossômico pelo exame de cariótipo;
- Não existe tratamento específico para SE, sendo realizado reanimação neonatal de forma individualizada e manejo das complicações naqueles que sobreviverem.
Definição da doença
Descrita pela primeira vez em 1960 por Edwards et al, a síndrome de Edwards (SE) é um distúrbio cromossômico autossômico devido a uma cópia extra do cromossomo 18, por isso é também chamada de síndrome da trissomia 18.
Epidemiologia e fisiopatologia da síndrome de Edwards
A SE é a segunda trissomia autossômica mais frequentemente observada ao nascimento, ficando atrás apenas da síndrome de Down, com prevalência em nascidos vivos variando de 1 em 3.600 a 1 em 10.000. Esta síndrome genética resulta de uma cópia extra do cromossomo 18, que pode ser completa, parcial e em mosaico.
A trissomia 18 completa é a forma mais comum (94%) em que cada célula contém três cópias completas do cromossomo 18. O mosaico é o segundo tipo mais comum, representando uma trissomia 18 completa e uma linha celular normal. Por fim, na trissomia parcial apenas um segmento parcial do cromossomo 18q tem presença triplicada, geralmente resultante de uma translocação ou inversão balanceada realizada por um dos pais.
Manifestações clínicas da síndrome de Edwards
Mais de 100 características clínicas foram associadas à síndrome de Edwards, mas nenhuma das características clínicas é patognomônica da doença. Alguns desses achados são descritos a seguir:
- Achados neurológicos: hipoplasia cerebelar, holoprosen, meningoencefalocele, anencefalia, hidrocefalia, deficiência mental grave, hipotonia neonatal seguida de hipertonia, retardo no desenvolvimento neuropsicomotor, apneia, convulsões.
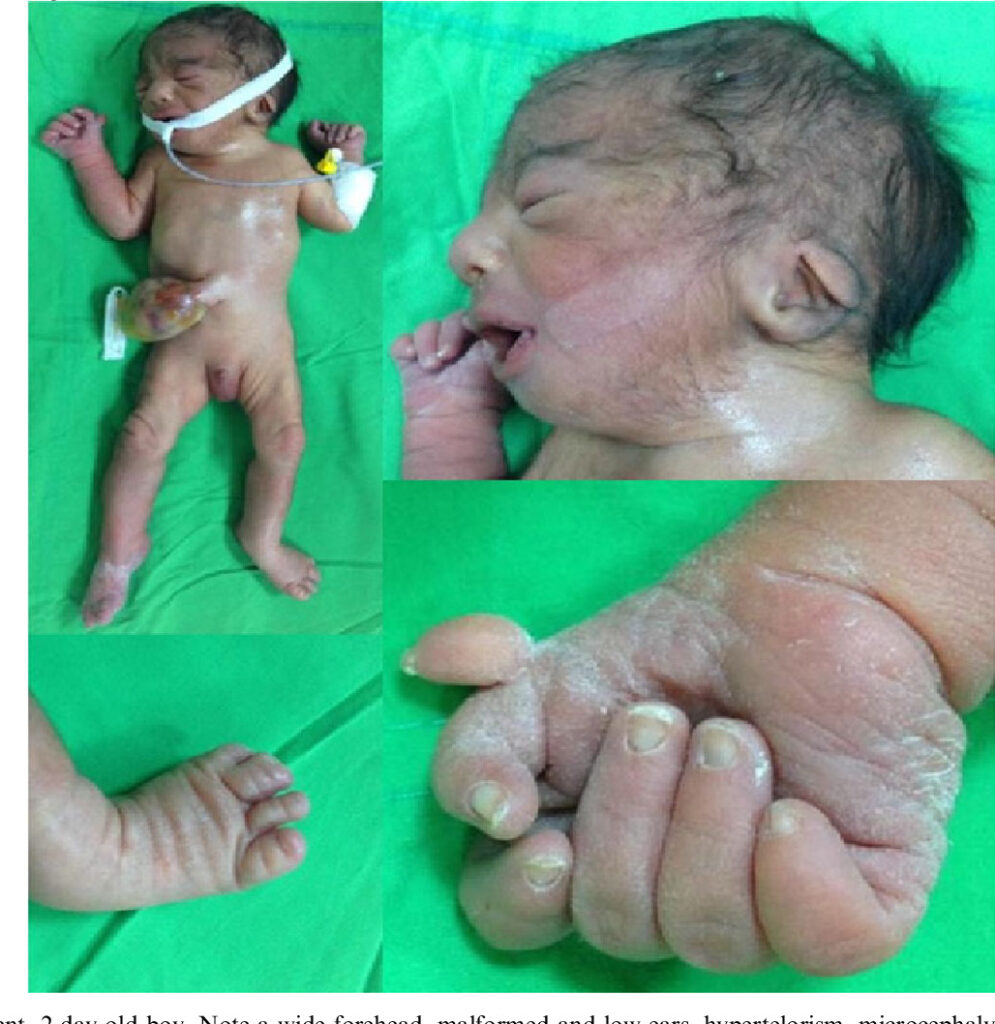
- Achados de crânio e face: Crânio dismórfico (microcefalia, estreitamento bitemporal e occipital proeminente), face triangular e assimétrica, orelhas com implantação baixa, microftalmia e hipoplasia das pálpebras.
- Achados em tórax: Hipoplasia pulmonar, com tórax relativamente largo, com ou sem hipertelorismo mamário, traqueobroncomalácia e laringomalácia.
- Achados em abdome: Onfalocele, atresia de esôfago com fístula traqueoesofágica, estenose pilórica, atresia ileal, má rotação, divertículo de Meckel, diástase abdominal.
- Defeitos cardíacos: A maioria dos pacientes tem defeito do septo ventricular ou atrial, persistência do canal arterial, tetralogia de Fallot, sobrecarga da aorta, coarctação da aorta e síndrome do coração esquerdo hipoplásico e doença cardíaca polivalvar.
- Extremidades: Tipicamente, os punhos estão cerrados, com o segundo dedo sobre o médio e o quinto sobre o quarto. A prega distal do quinto dedo pode estar ausente e, menos frequentemente, pode ocorrer ausência das pregas distais do terceiro e quarto dedos. Dedos supranumerários também são comuns.
Um pequeno número de bebês nascidos vivos com a síndrome de Edwards viverá após o primeiro ano de vida. A principal causa de morte súbita na síndrome de Edwards é a instabilidade neurológica, insuficiência cardíaca e insuficiência respiratória.
Diagnóstico da síndrome de Edwards
A SE é diagnosticada a partir da percepção clínica, geralmente ao nascimento, raramente confundido com outras condições. É importante lembrar que o diagnóstico da SE é usualmente confirmado por meio do estudo cromossômico pelo exame de cariótipo ao evidenciar uma trissomia completa ou parcial do cromossomo 18.
A triagem de cromossomopatias começa na USG morfológica do 1° trimestre em busca do aumento da translucência nucal. A amniocentese ou biópsia de vilosidades coriônicas são recomendadas se a triagem pré-natal sugerir um alto risco de aneuploidia fetal.
Estudos complementares devem ser realizados para avaliar anormalidades intracranianas (ressonância magnética), cardíacas (ecocardiograma), intra-abdominais e renais (ultrassonografia).
Tratamento da síndrome de Edwards
Como já vimos, a maioria dos fetos portadores de trissomia do cromossomo 18 não chega ao termo, ou seja, acaba indo a óbito durante a vida embrionária e fetal. Quando nascidos vivos, não há opção de tratamento definitivo para a síndrome de Edwards.
#Ponto importante: Algumas diretrizes internacionais, como o Programa de Ressuscitação Neonatal da Sociedade Americana, não estão mais defendendo os esforços de ressuscitação na sala de parto de pacientes com SE. No Brasil, é necessário ter a comprovação diagnóstica antenatal e considerar a vontade dos pais e os avanços terapêuticos existentes para decidir quanto à conduta em sala de parto.
A cirurgia cardíaca paliativa e corretiva raramente é realizada, mas pode ser feita para defeitos cardíacos congênitos complexos. Para aqueles com problemas gastrointestinais, a alimentação por sonda nasogástrica ou por gastrostomia são consideradas para resolver os problemas de alimentação. As infecções devem ser tratadas semelhante a abordagem padrão.

Veja também:
- Resumo de mielomeningocele: diagnóstico, tratamento e mais!
- Resumo de doenças gastrointestinais do recém-nascido: diagnóstico, tratamento e mais!
- Resumo de cardiopatias congênitas: diagnóstico, tratamento e mais!
- Resumo de baixa estatura: avaliação do crescimento, etiologias e mais!
- Resumo de marcos do desenvolvimento infantil: diagnóstico, tratamento e mais!
- Resumo da síndrome do desconforto respiratório agudo no recém-nascido: diagnóstico, tratamento e mais!
Referências bibliográficas:
- Balasundaram P, Avulakunta ID. Edwards Syndrome. [Updated 2023 Mar 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK570597/
- Demir N, Doğan M, Peker E, Bulan K, Tuncer O. A very rare entity of diabetes insipidus associated with Edwards syndrome. Genet Res (Camb). 2013 Aug;95(4):130-2. doi: 10.1017/S0016672313000165. PMID: 24074370.https://pubmed.ncbi.nlm.nih.gov/24074370/
- Rafael Fabiano M. Rosa et al. Trissomia 18: revisão dos aspectos clínicos, etiológicos, prognósticos e éticos. Rev Paul Pediatr 2013;31(1):111-20. https://www.scielo.br/j/rpp/a/dsM36zFXP5VnNb7Z4kyjsns/?format=pdf&lang=pt
- Crédito da imagem em destaque: Pexels